One of the most important things when we study a crystal or a surface, is to have a nice and relax geometry, that means that you have to find the geometry with lowest energy. To do that, eventhough you start from geometries that comes from experimental data, you have to perform a geometry relaxation.
Remember that we are studying periodic systems, so we have two very important concepts: the crystal unitcells and the possition of ions, both becomes in the crystal structure. We can make a distinction between the unitcell:
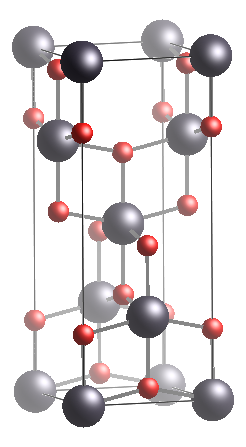
Figure 1. Titanium Dioxide Anatase phase. Left: Unitcell. Right: Ion Possition whithin the unitcell.
Types of relaxation in VASP
VASP allows you to make different types of relaxation:
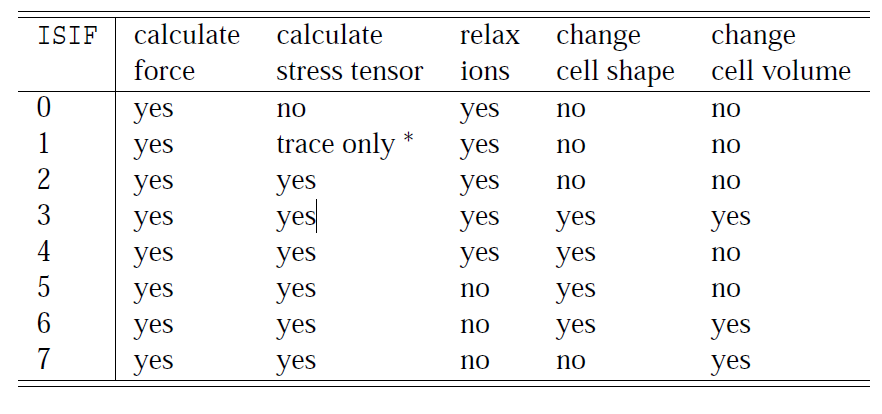
The most usally used are:
ISIF=2: Relax
ISIF=3: Full Relaxation.
ISIF=0 ; NSW=0: Performs a single point calculation, no stress tensor calculation.
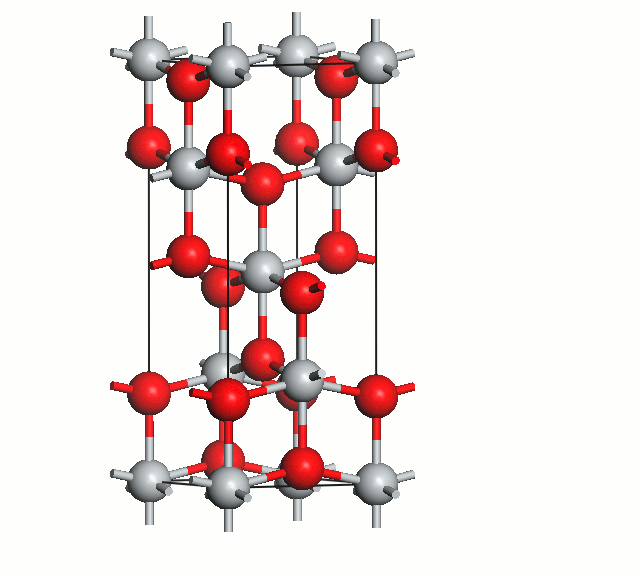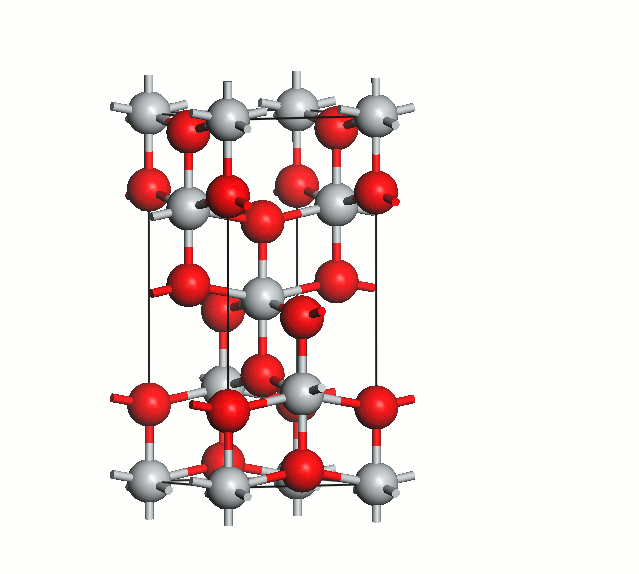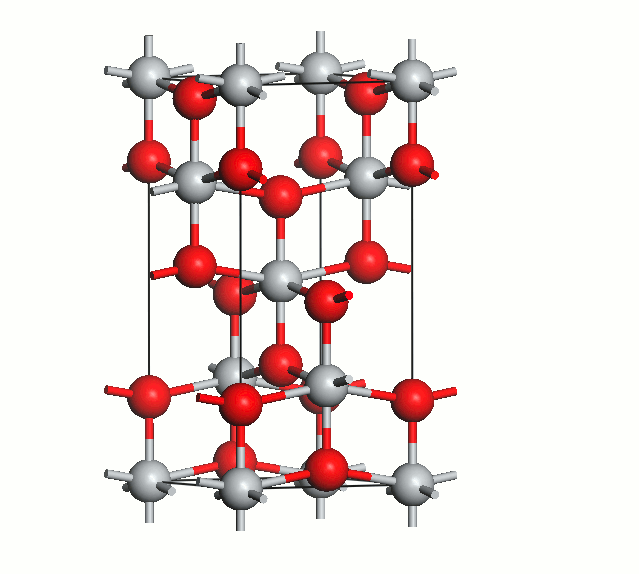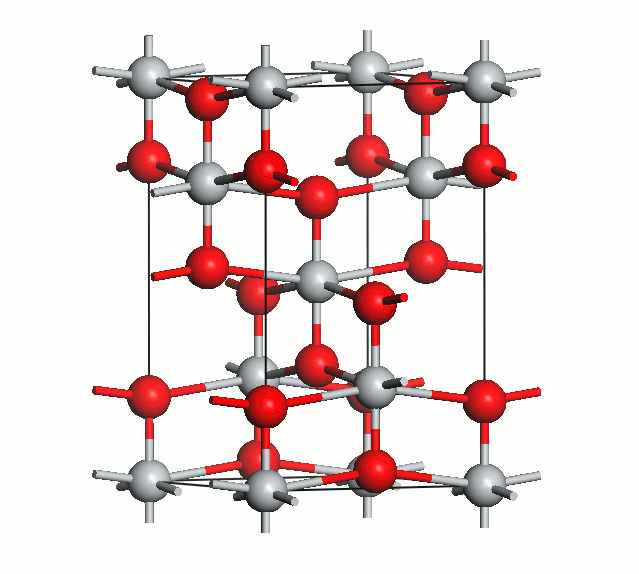
Figure 2. Anatase cell relaxation.
Full Relaxation
To perform a full geometry optimization first prepare the POSCAR file. In this example we are going to relax the anatase structure.
- Download the .cif structure from the net.
- Open the structure file with VESTA software.
- Analize the structure and export the structure into a POSCAR type file.
You should see something like below:
Anatase full geometry relaxation
1
3.78500 0.00000 0.00000
0.00000 3.78500 0.00000
0.00000 0.00000 3.78500
Ti O
4 8
Direct
0.000000000 0.000000000 0.000000000
0.500000000 0.500000000 0.500000000
0.000000000 0.500000000 0.250000000
0.500000000 0.000000000 0.750000000
0.000000000 0.000000000 0.206599995
0.500000000 0.500000000 0.706600010
0.000000000 0.500000000 0.456600010
0.500000000 0.000000000 0.956600010
0.500000000 0.000000000 0.543399990
0.000000000 0.500000000 0.043400005
0.500000000 0.500000000 0.293399990
0.000000000 0.000000000 0.793399990
INCAR preparation
The INCAR file, as told it before, is the file with the main directives.
In this case we have to perform a full relaxation, that is to say ion
possitions and cell relaxation. So the important flags are:
- ISTART= 0 New Job.
- ISIF = 3 Cell and ions relaxation.
- IBRION= 2 DFT Conjugate Gradient.
- NSW = 25 Maximum Optimization steps.
- EDIFF & EDIFFG Convergence Criteria.
Some remarks:
-ENCUT is the max kinetic energy cutoff of all atoms that appears on the
POTCAR file. In this case oxygen atom has the greatest encut 400 eV.
-ICHARG=2 the initial charge distribution are the atomic charges.
# Full relaxation cell+ion Anatase volume #=========================================== # General Setup System = Anatase # Calculation Title PREC = NORMAL # Options: Normal, Medium, High, Low ENCUT = 400 # Kinetic Energy Cutoff in eV ISTART = 0 # Job: 0-new 1-cont 2-samecut ICHARG = 2 # initial charge density: 1-file 2-atom 10-cons 11-DOS ISPIN = 1 # Spin Polarize: 1-No 2-Yes # Electronic Relaxation (SCF) NELM = 60 # Max Number of Elec Self Cons Steps NELMIN = 2 # Min Number of ESC steps NELMDL = 10 # Number of non-SC at the beginning EDIFF = 1.0E-05 # Stopping criteria for ESC LREAL = .FALSE. # Real space projection IALGO = 48 # Electronic algorithm minimization VOSKOWN = 1 # 1- uses VWN exact correlation ADDGRID = .TRUE. # Improve the grid accuracy # Ionic Relaxation EDIFFG = -1.0E-04 # Stopping criteria for ionic self cons steps NSW = 25 # Max Number of ISC steps: 0- Single Point IBRION = 2 # Ionic Relaxation Method: 0-MD 1-qNewton-RaphsonElectronic 2-CG ISIF = 3 # Stress and Relaxation: 2-Ion 3-cell+ion ADDGRID = .TRUE. # Improve the grid accuracy SIGMA = 0.10 # Insulators/semiconductors=0.1 metals=0.05 ISMEAR = 0 # Partial Occupancies for each Orbital # -5 DOS, -2 from file, -1 Fermi Smear, 0 Gaussian Smear # Parallelization NPAR=8
POTCAR
We have to combine the potential files of Ti and O. To do that copy the POTCAR files into your work directory with diferent name. Potential files are in the directory where you install VASP. Finally just concatenate both files into a new file called POTCAR. Remember conserve the order that we impose in the POSCAR.
[user@machine WRKDIR]$ cp ~/VASP/vasp5.x/Potential/PBE/Ti_sv/POTCAR POTCAR_Ti
[user@machine WRKDIR]$ cp ~/VASP/vasp5.x/Potential/PBE/O/POTCAR POTCAR_O
[user@machine WRKDIR]$ cat POTCAR_Ti POTCAR_O >> POTCAR
KPOINTS file
We are going to generate an automatic K-mesh with 216 K-points distributed in all directions.
Automatic Mesh # Generates Automatically the K-mesh 0 Monkhorst-pack # Automatic Distribution of the mesh 6 6 6
Job administrator
If you have a queue administrator you have to set the queue file. FamaLab has torque as default job manager. Please paste the queue file .pbs or .sge into your work directory.
The file has as main directives the jobname, the number of cpu you are requesting, the time is going to take your calculation, and which of the vasp programs you will use.
#PBS -N name_identifier #PBS -o vasp.out #PBS -j oe #PBS -e vasp.err #PBS -l walltime=8760:00:00 #PBS -l nodes=1:ppn=8 #PBS -q batch #Elegir la version de vasp: VASP=vasp_complex #Volume=vasp_complex, supercell=vasp_cd, molecules=vasp_gamma
The variables you can modify are marked in red:
- Name identifier is the label on the queue of the jobs, it cannot start with a number.
- The ppn variable is the number of cores (cpu's) you specify on the INCAR.
- vasp_complex: is used when the system has all symmetries. For volume structures.
KPOINTS N N N
- vasp_cd: is for a systems which has only two periodic dimensions.
For surfaces. KPOINTS N N 1.
- vasp_gamma: is fot system with non periodicity. For molecules. KPOINTS N 1 1
You can download the build files here:
INCAR
POTCAR
KPOINTS
POSCAR