Index
|
Input File
Orca is a very friendly software that allow to do several types of calculus with a few lines. Almos all flags can be extended to modify different parameters, it is all documented on the orca manual
Method
Orca can perform serveral methods:
- HF Hartree-Fock
- MP2, MP3, Coupled Cluster, CAS, MRCI
- DFT
- MD
Each methods has his own flags to specify some parameters.
Runtype
The runtype can be combine with the methods (check compatibility) above, some of the most relevants are.
| ENERGY |
Single Point Calculation |
| OPT |
Geometry Optimization |
| ENGRAD |
Energy Gradient Calculation |
| SCAN |
Scan for Geometric parameters |
Basis Set
Orca has available the Slater Type Orbitals and the Gaussian Type Orbital basis sets, among others, here you can find a list of all basis sets.
Convergence on the SCF
SCF convergence is a pressing problem in any electronic structure package because the total execution times increases linearly with the number of iterations. ORCA has implemented tools to balance precision and computation time.
- Convergence tolerance
- Dynamic and Static Damping
- Level Shifting
- Direct Inversion in Iterative Space (DIIS)
- Approximate Second Order SCF
Coordinates
There are two different ways to specify the possition of the atoms, with the cartesian coordinates or with the Z-matrix coordinates. Both do geometry optimizations (with or without constraints) and scan potential surface paths.
To specify an the cartesian format you have to enter the Atomic simbol and the xyz coordinates.
Charge Population and Bond Orders
Hirshfeld Charge Population and Bond Orders are two handly tools to analyze molecular systems, to print this properties, you have to add the next lines:
%output
Print [ P_Hirshfeld ] 1
Print[ P_Mayer ] 1
end
Other important Flags
Direct - To save some computing time, not all integrals are stored on the hard drive, instead orca recomputes all two-electron integrals in each SCF.
XC-Functional - If the DFT method is used, you have to specify the exchange-correlation functional. All XC functionals can be found it here.
Pmodel - It consists of building and diagonalizing a Kohn-Sham matrix (as an initial guess) with an electron density which consists of the superposition of spherical neutral atoms densities.
|
Input Examples
|
PBS file (only for torque users)
To make our calculation on several CPU's we have tu use the *.pbs file. This file has the instructions about running the calculation, and once it's done very few things have to change for the other calculations.
Here at the FAMAlab we have a script which copy the input files and paste it on a temporal allocation where the calculation is done. This copy and paste files is done to run the calculation on a faster SSD which reduces the computing time. You can see the script here.
Next flagas allow you to manipulate the pbs script.
#PBS -N name Name to idenfity the work on the queue.Be sure that name starts with an alphabetic character
#PBS -l walltime=hh/mm/ss Lenght time for the calculation. If the computer time exceds this value, torque will stop the job.
#PBS -l nodes=n ppn=cpus Specifies the amoutn of nodes and the cpu's per nodes fot the job.
FILE=input_file N ame of the orca input file with no extension.
|
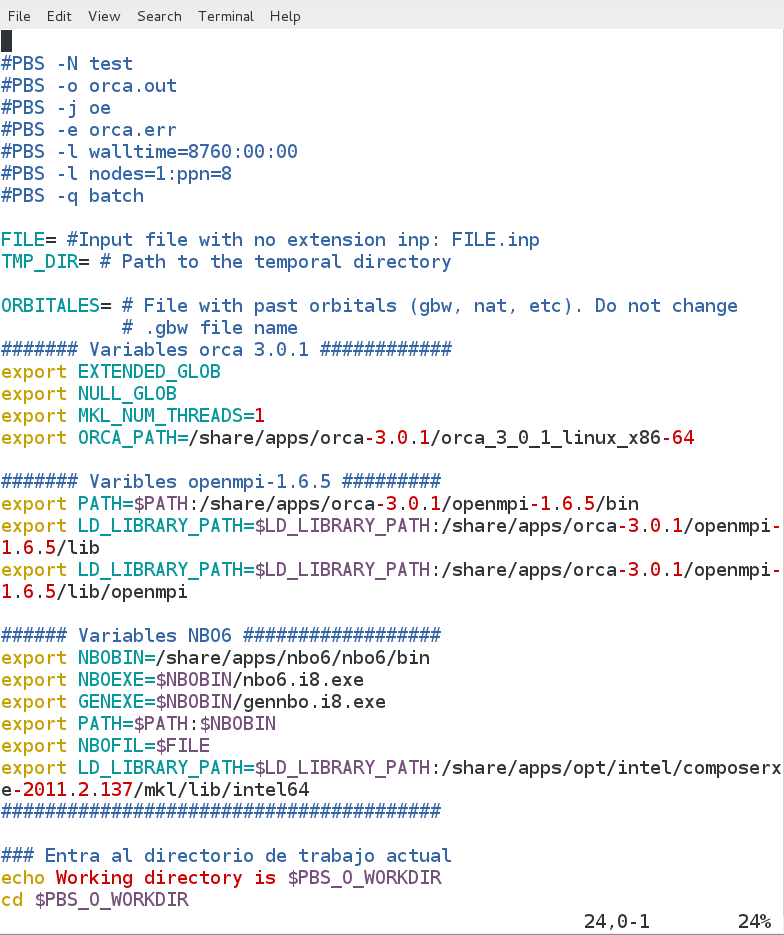
orca-3.0.1.pbs file |
|
